Today we will pick up where we left off last week and perform the following steps:
Trim Illumina adapters from our reads
Start by uploading the paired end reads for the file we were exploring last week. You can fetch the fastq file here. You should now have the following fastq files available on Galaxy:
SRR2584863_1.trim.sub.fastq
SRR2584863_2.trim.sub.fastq
Trimming
Open the Trimmomatic tool on Galaxy.
Trimmomatic tool on Galaxy
Select the following inputs and parameters for Trimmomatic:
Singe-end or paired-end reads: paired-end
Input FASTQ file 1: SRR2584863_1.trim.sub.fastq
Input FASTQ file 2: SRR2584863_2.trim.sub.fastq
Perform Illuminaclip step: Yes
Standard adapter sequences
Adapter sequences: Nextera (paired-end)
Keep all other defaults
Trimmomatic operation:
Sliding window trimming
Number of bases to average across: 4
Average quality required: 20
Trimmomatic operation (insert a second operation):
MINLEN: 25
Keep all other defaults
Rerun FastQC to verify that the trim step was successful.
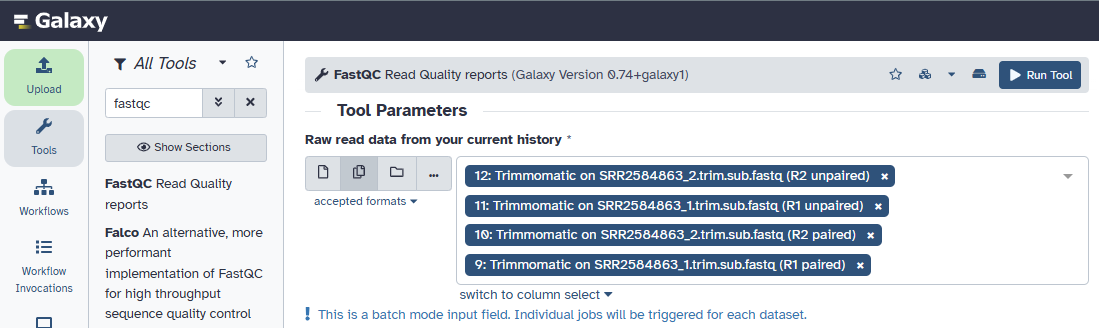
We now have 4 files to run. Select the multiple datasets option for the input field and select the output files from Trimmomatic.
FastQC tool on Galaxy
QC
The unpaired read files are going to be very small and hard to tell what is going on with any reliability, but the two paired files still don’t look quite right.
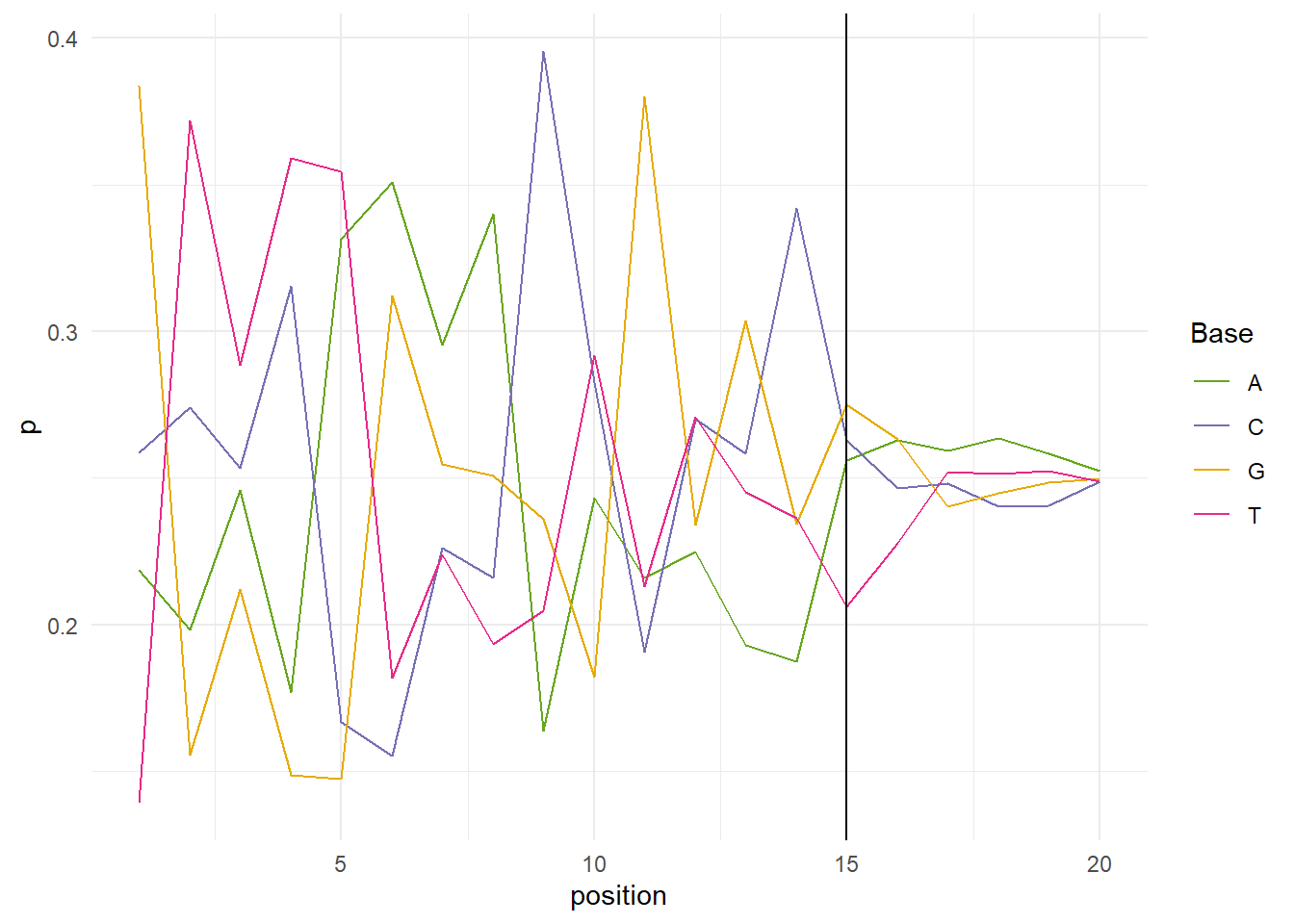
A warning is thrown by FastQC if the difference between two bases is greater than 10%, and an error is thrown if the difference is greater than 20%. If we look at the per sequence content plot of the first 20 positions of SRR254863_1, it looks like we should be find trimming the head of our sequences up to 15 base pairs.
Code
library(ShortRead) # install with BiocManager::install("ShortRead")library(stringr)library(tidyr)library(ggplot2)library(RColorBrewer)root <- here::here()# read sequencesreads <-readFastq(file.path(root, 'data', 'ngs', 'SRR2584863_1.trim.sub.fastq')) |>sread()# split out nucleotides in the first 20 positionsnbp <-20bp_by_pos <-subseq(reads, start =1, end = nbp) |>as.character() |>str_split('') |>unlist() |>matrix(ncol = nbp, byrow =TRUE)# set up our own per base sequence content plotpb <-tibble(position =1:nbp,A =apply(bp_by_pos =='A', 2, mean),C =apply(bp_by_pos =='C', 2, mean),`T`=apply(bp_by_pos =='T', 2, mean),G =apply(bp_by_pos =='G', 2, mean)) |>pivot_longer(cols =2:5, names_to ='Base', values_to ='p')ggplot(pb, aes(position, p, color = Base)) +geom_line() +theme_minimal() +geom_vline(xintercept =15) +scale_color_manual(values =brewer.pal(n =7, name ='Dark2')[c(5,3,6,4)])
Per base sequence content over the first 20 positions of SRR2584863_1
Run Trimmomatic again using the settings as above plus an additional Trimmomatic operation:
HEADCROP: 15
Rerun FastQC on the 2 pared files created by Trimmomatic (we’ll skip the unpaired files this time).
Read mapping
Once we are happy with our QC report, let’s move on to mapping the reads to a reference genome and calling some variants!
Open the BWA-MEM2 tool in Galaxy and select the following as inputs:
Trimmomatic on SRR2584863_1.trim.sub.fastq (R1 paired)
Trimmomatic on SRR2584863_2.trim.sub.fastq (R2 paired)
BWA-MEM2 tool on Galaxy
Variant calling
Open the bcftools mpileup tool and select the following inputs:
Input: xx: BWA-MEM2 on data yy and data zz (mapped reads in BAM format) where xx and yy correspond to the job numbers for the different steps we’ve run thus far.
Use the same locally cached Ecoli reference genome we used above: Escherichia coli O157:H7 str. Sakai (GCF_000008865.2)
bcftools mpileup tool on Galaxy
Open the bcftools call tool and select the following inputs:
VCF/BCF data: xx: bcftools mpileup on data yy where xx and yy correspond to the job ids for the previous two steps.
Calling method: consensus caller
bcftools call tool on Galaxy
Annotation
Open the bcftools csq tool and select the following inputs:
VCF/BCF data: xx: bcftools call on data yy where xx and yy are the job ids for the previous two steps.
Reference genome: Escherichia coli O157:H7 str. Sakai (GCF_000008865.2)
GFF3 annoation file: GCF_000008865.2_ASM886v2_genomic.gff.gz (this can also be found on the corresponding NCBI page under FTP)
bcftools csq tool on Galaxy
I had trouble getting this to work, so let’s try a different approach. This will take longer to run, since we aren’t using a pre-annotated gff file.
Convert your bcf file to a fasta file with the bcftools consensus tool. Galaxy should remember your reference genome.
bcftools consensus tool on Galaxy
Annotate the consensus sequence with the Bakta tool - use the defaults.
Bakta tool on Galaxy
This gives us several outputs (see Ensembl for additional information):
Summary: I think this can be ignored, as it seems to be identical to the gff file. There are some visualization tools you could use to look at the information in the gff file, but these visualizations are rudimentary.
gff: The gff file contains annotations for our sample’s genome. Columns include:
Contig where the feature was found
Database where the annotations come from
Type of annotation, for example: CDS (coding DNA sequence), ncRNA (non-coding RNA), regulatory region
Start and end positions of the region
Score: E-value for the feature (if available)
Strand (+/-)
Phase: indicates the first base of the reading frame where
0: feature starts on the first base of a codon
1: feature starts on the second base of a codon
2: feature starts on the third base of a codon
Metadata with additional information regarding the region
A fasta file with all nucleotide sequences for each feature in the gff file
A ciros plot of the data with one section per contig. The location of features is shown in the outer rings and the gc content (red/green) and gc content skew (blue/gold) are shown in the inner rings.