Acknowledgments
Gemini and NotebookLM were used to find and summarize reference material included in these notes.
Introduction
- Structural Bioinformatics specifically focuses on
Protein structure
Proteins are essential molecules performing vital roles
- Providing structural scaffolding
- Participating in metabolism
- Biosignaling
- Gene regulation
The three-dimensional (3D) structure of a protein is fundamentally linked to its function
Understanding the structure provides insight into protein function
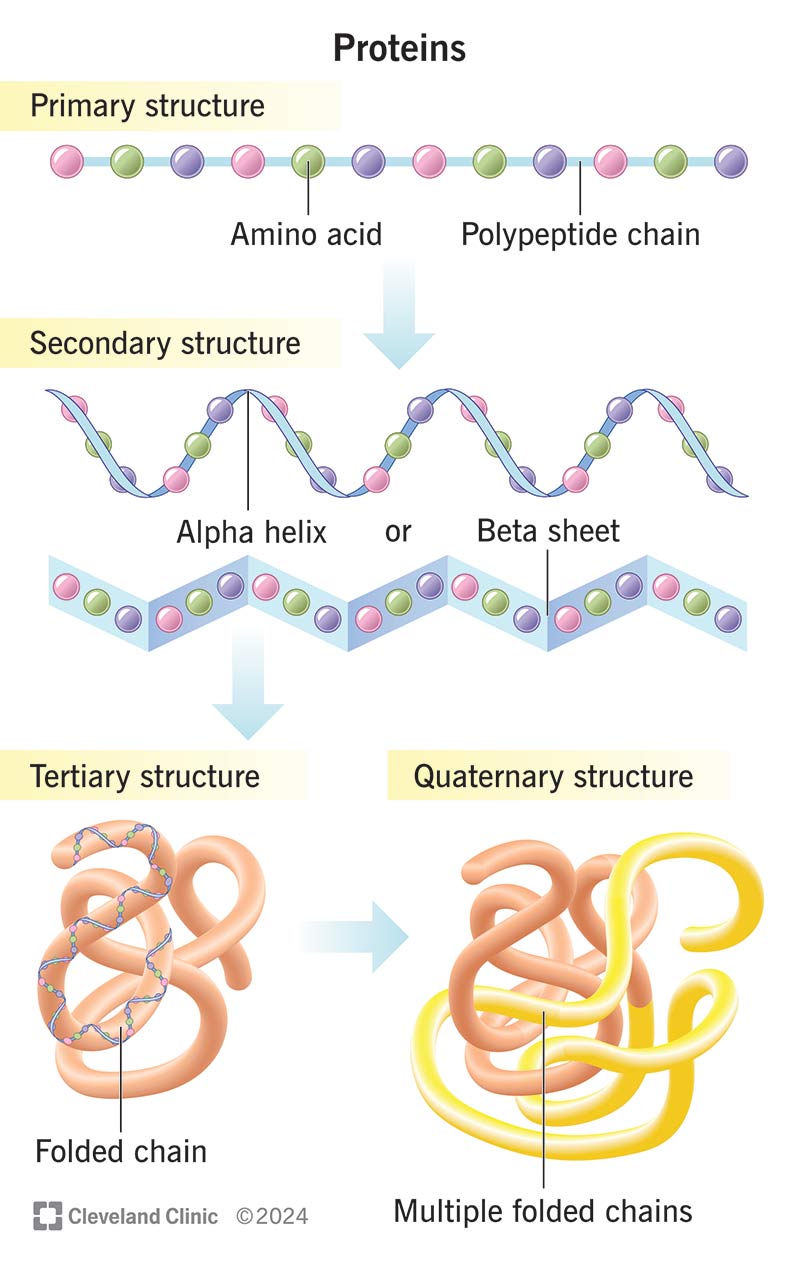
Protein Structure Levels
- Primary structure
- The unique sequence of amino acids
- Secondary structure
- Local folding patterns, typically comprising \(\alpha\)-helices and \(\beta\)-sheets
- Loop and turn regions link these structures
- 3D structure (tertiary/quaternary)
- Complete folding pattern of the protein
- Essential for analyzing active sites and investigating molecular interactions
Protein Structure Databases and Visualization
Primary structure databases
- GenBank (Genetic Sequence Databank)
- Nucleotide sequences with a flat file structure
- Files are ASCII text files that can be read by humans and computers
- Contain data such as accession numbers, gene names, and phylogenetic classification
- GenBank is maintained by the NCBI
Secondary structure databases
- UniProt (Universal Protein Resource)
- Protein sequences
- Functional annotations
- Structural details
- Swiss-Prot
- Reviewed and manually annotated section of UniProt
- Higher quality data
- Recognized by a golden star in UniProt
3D structure databases
- Protein Data Bank (PDB)
- Database for 3D structures of biological macromolecules, including proteins and nucleic acids
- Typically experimentally determined using methods like X-ray crystallography or NMR spectroscopy
- Hosted by the Research Collaboratory for Structural Bioinformatics (RCSB).
Structure quality and validation
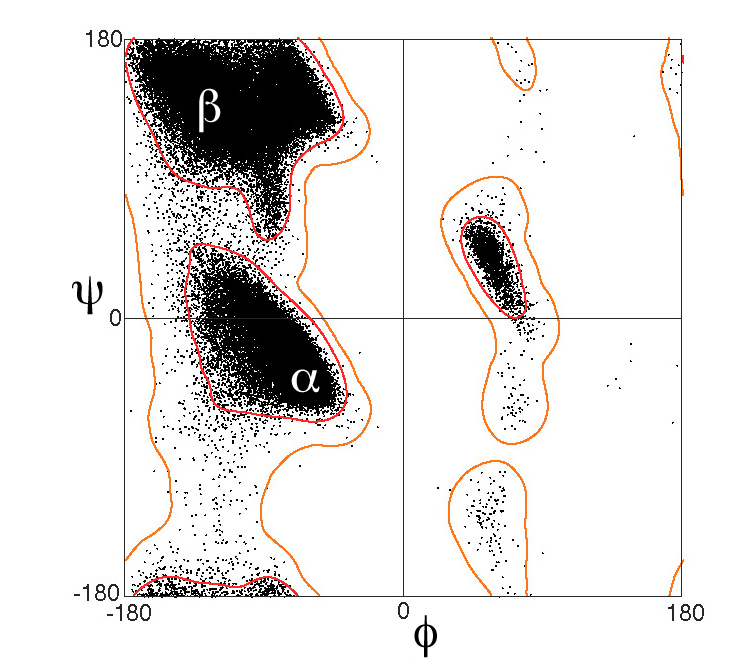
- Ramachandran plot
- Tool used to check the quality of protein structures
- This plot validates the main chain of amino acids by comparing observed versus expected \(\phi\) (phi) and \(\psi\) (psi) angles, helping to identify potential errors or stereochemical impediments
- The Bumbling Biochemist has a good video discussing these (the bumbling biochemist 2023)
- Structure Visualization Tools
- PyMOL
- Tool for protein modeling
- Swiss-PdbViewer (DeepView):
- Analysis of multiple proteins simultaneously
- Includes superposition
- Can also be used to examine the Ramachandran plot
Computational Protein Modeling Methods
Only a fraction of known proteins have had their 3D structure solved experimentally, which is costly and time-consuming
Computational methods are critical for most porteins for
- Functional understanding
- Drug discovery
Modeling strategies
- Template-Based Modeling (TBM)
- Builds the structure of a target protein based on the known 3D structure (template) of a homologous protein
- Assumpes that proteins with similar sequences fold into similar structures
- Most effective when the target and template share more than 30% to 40% sequence similarity
- De novo modeling (Ab Initio)
- Used when a suitable template structure is not available
- Estimates protein’s structure from scratch
- Calculates the most energetically favorable conformation based on chemical and physical principles
TBM prediction process
- Reference Identification:
- Identify similar amino acid sequences whose structures are resolved
- Often using tools like BLAST
- Template Selection:
- Choose one or more suitable template structures
- Consider factors like sharing the same function or belonging to the same protein family
- Alignment:
- Align the target and template sequences
- Alignment algorithms use
- Scoring matrices like BLOSUM and PAM
- Gap penalties for insertions or deletions
- Construction:
- Transfer the template’s coordinates to the target protein to generate the atomic coordinates of the 3D model
- Model Validation:
- Verify the quality of the model by checking for errors, such as
- Unsuitable torsion angles
- Steric hindrance
- Ramachandran plots can be helpful
Modeling Interactions and Functional Prediction
Protein-protein interaction (PPI) modeling
Docking methods for PPIs
- Docking is used to model PPIs, predicting the 3D structure of protein complexes
- Rigid Body Docking assumes that proteins maintain their structure upon binding
- Energy-Based Docking employs molecular mechanics force fields and scoring functions to estimate the binding free energy
- Machine learning algorithms can be trained on known PPI data to predict binding affinities and accelerate simulations.
Molecular dynamics (MD) simulations
Provide a thorough perspective on protein movement, dynamics, and conformational changes during binding
Atomistic MD Simulations mimic the behavior of individual atoms over time using classical force fields
Coarse-Grained (CG) MD Simulations
- Simplify the molecular representation to reduce computational complexity
- Allows for the simulation of larger complexes over longer timescales
Interaction databases
- STRING (Search Tool for the Retrieval of Interacting Genes/Proteins):
- Provides details regarding quality-controlled protein-protein association networks
- MINT (Molecular INTeraction Database):
- A repository containing experimentally supported PPIs
- Cytoscape:
- Open-source software used for visualizing and analyzing intricate PPI networks
Functional annotation and pathway analysis
- Gene Ontology (GO):
- Hierarchical, standardized vocabulary describing protein functions across three categories:
- Cellular Component
- Biological Process
- Molecular Function
- InterPro: Combines data from several sources to predict
- Protein domains
- Families
- Functional sites by combining data from several sources (e.g., Pfam, PROSITE)
- KEGG (Kyoto Encyclopedia of Genes and Genomes):
- Database providing pathway maps and combining genetic, functional, and chemical domains
- ScanNet:
- A geometric deep-learning model designed to learn features directly from protein structures
- Can be used to predict functional sites, such as binding sites for small molecules
Summary
- Bioinformatics facilitates
- Structure prediction
- Simulating interactions
- Clarifying functional relationships
- Investigation of protein-protein interactions
- Understanding function
- Predictive accuracy of protein structure, exemplified by tools like AlphaFold, is rapidly advancing
Future Directions
- AI, AI, AI …
- DL models capable of integrating information across molecular, cellular, tissue, and organism levels is expected to lead to a more complete understanding of biological processes
- Research direction: Integrating multi-omics data
the bumbling biochemist. 2023.
“Understanding & Exploring & Ramachandran Plots & Phi/Psi Angles w/o Getting Bogged down in Details.” https://www.youtube.com/watch?v=QGBxXHqdC9E.