15 Metagenomics
Acknolegments
NotebookLM, Perplexity and Google were used for collecting and summarizing references while preparing these lecture notes.
What is Metagenomics?
Metagenomics: the analysis of all genomes (microbiome) from all microbiota in a sample
High-throughput sequencing of genetic material recovered directly from environmental samples
Like all high-throughput technologies, metagenomics has revolutionized microbiology
Applications to human medicine (e.g. the human microbiome)
Targeted Approach: 16S rRNA Amplicon Sequencing
16S rRNA amplicon sequencing
Offers quick, cheap sequencing solution to characterize all microbes in a sample (microbiome)
Allows annotation using extremely detailed and well-curated taxonomic databases
DNA is extracted from the environmental sample (e.g. fecal samples)
PCR is used to amplify a variable region (e.g. V4-V5) using primers constructed with adaptors and barcodes
16S rRNA

Entire gene is ~1500 bp long
Many regions are highly conserved across all prokaryotes
Forms the backbone of ribosomes
Encode the structure needed for translation of mRNA to protein
Other regions (V1-V9) are hypervariable and correlate strongly with taxonomy (Gray, Sankoff, and Cedergren 1984; Yang, Wang, and Qian 2016)
- Vary in length from 10 to 100 bp
16S bioinformatic pipeline
Major software platforms used for analysis include
Quality control and alignment
- Raw data undergoes quality trimming to remove low-quality bases
- Pairing of forward and reverse reads into contigs
- Highly conserved regions provide a strong anchor

Removal of artifacts
- Chimeras (biased merged sequences from two origins) are identified and removed, as they artificially increase diversity
- Mitochondrial and chloroplast DNA may be filtered
- DNA from other domains (e.g. human) are filtered
Grouping reads
- Reads are grouped into Operational Taxonomic Units (OTUs)
- Typically clustered using a 97% similarity threshold
- Corresponds to the taxonomic threshold between prokaryotic species
Grouping reads
Taxonomic Assignment
- OTUs are aligned against specialized 16S rRNA gene sequence databases for annotation
- SILVA
- Greengenes
- RDP
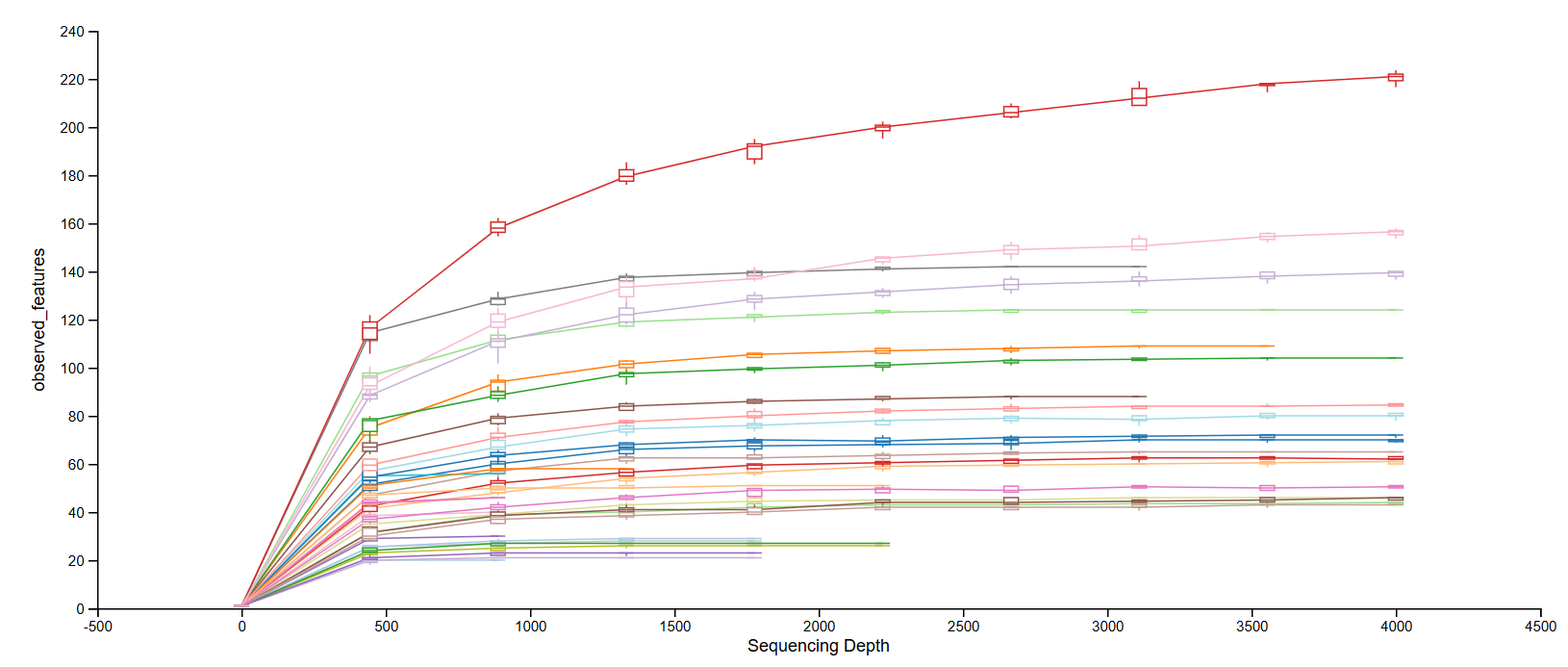
Alpha (α)-Diversity
Measures diversity within a sample group
Rarefaction analysis explores if the sequencing depth was sufficient to capture the true diversity (plateauing curve indicates sufficiency)
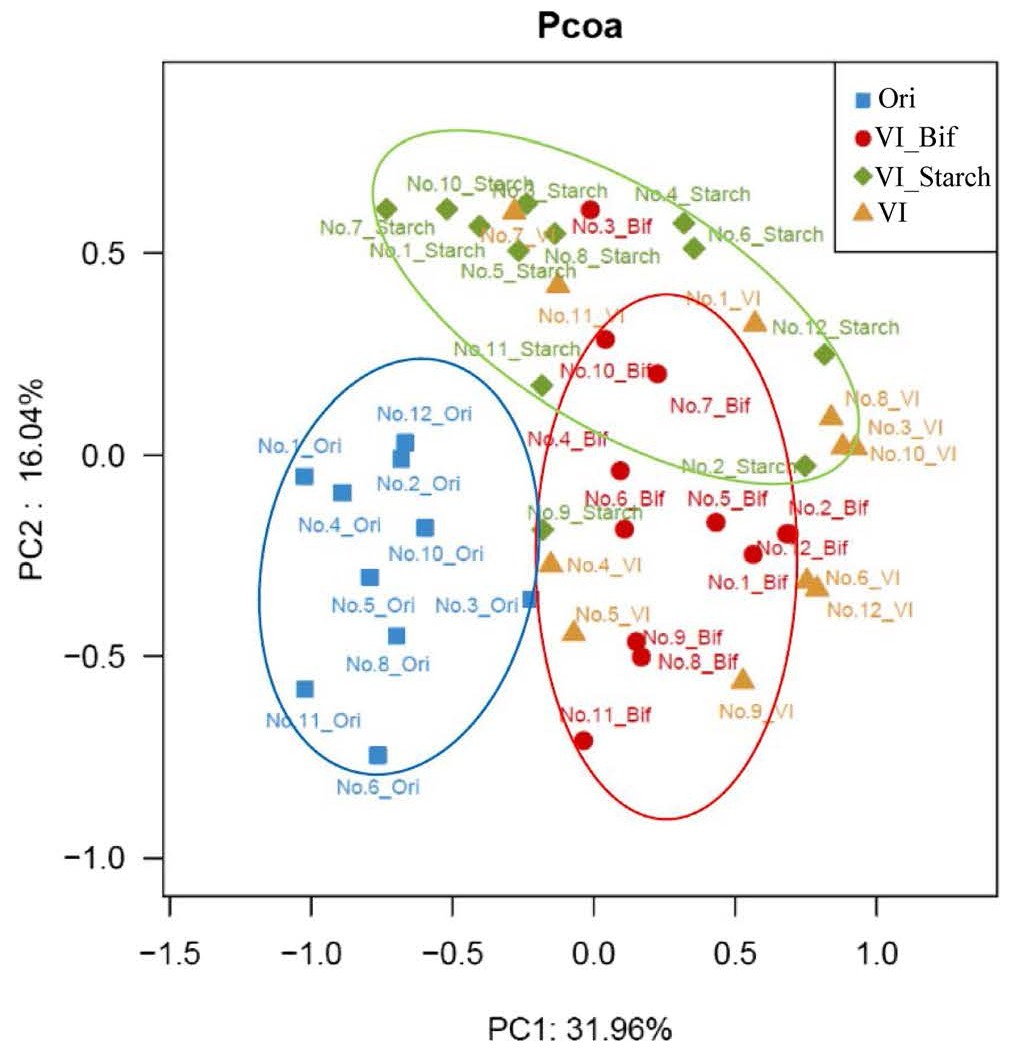
Beta (β)-Diversity
- Measures diversity between groups (similarity/dissimilarity comparison)


- Principal Coordinates Analysis (PCoA) can be used to observe sample clustering and differences
- Dimensionality reduction using dissimilarity matrix as input
- Different from traditional Principal Component Analysis (PCA) that analyzes features directly (e.g. protein abundance)
Comprehensive Approach: Full Shotgun DNA Metagenomics
Full DNA Shotgun Metagenomics sequences all DNA in a sample, aiming to reconstruct genome fragments (Metagenome-Assembled Genomes, MAGs)
Provides functional characteristics and allows investigation of the general diversity of all organisms
DNA extraction should yield at least 50% microbial DNA
Samples are sequenced using high-throughput platforms like Illumina or Nanopore
Pipeline for shotgun data
Preprocessing
Similar to 16S, reads are demultiplexed and subject to quality control
Removal of host DNA by mapping raw reads to the host genome for filtering (e.g. using tools like Bowtie 2)
Assembly
Environmental / microbiome samples are highly complex
Specialized assemblers are used (e.g. metaSPAdes, MEGAHIT)
Assess quality of the assemblies (e.g. CheckM)
Annotation (functional assignment)
Open Reading Frames (ORFs) that encode proteins are predicted
Functional annotation compares predicted proteins to databases like M5nr, SEED Subsystems, or KEGG
Sequence information linked to function, providing the functional potential of the community
Dedicated pipelines/servers
MG-RAST (Metagenomics RAST): User-friendly public resource that automates quality control, annotation, and comparative analysis against multiple databases. It uses BLAST or BLAT for searching.
MEGAN: Links taxonomy with function using the Lowest Common Ancestor (LCA) algorithm, often comparing against NCBI taxonomy
Prediction of function via taxonomy shortcut
- Tools like PICRUSt predict the abundance of gene families in microbial communities based on 16S rRNA marker gene sequences, providing a functional estimate without full shotgun sequencing
Single-Cell Techniques and Advanced Analysis
General similarity to regular scRNA-seq, but also has some unique challenges
No standardized methodologies yet (Ling et al. 2025; Gourlé et al. 2025)
DNA sequencing for single-cell sequencing follows the same principle as sequencing for single genomes
Specialized assemblers (like SPAdes) can handle single-cell genomes/mini-metagenomes
scRNA-seq provides high granularity, allowing full insight into the interplay of transcripts within individual cells
Challenges in scRNA-seq for metagenomics
Sparsity/dropout (similar to regular scRNA-seq)
- Measurements often have large fractions of observed zeros, referred to as “dropout”
- Combination of technical noise and the true biological absence of expression
- Sparsity hinders downstream analysis
Statistical Frameworks
- New statistical frameworks are needed to deal with the high granularity of changes and the uncertainty in clustering/cell type assignment prior to differential analysis
General computational techniques for single cell metagenomics
Data preprocessing and normalization
Necessary for deep-learning models and large datasets like metagenomics
Helps reduce the impact of noise and ensures comparability
Normalization methods include: total count, upper quartile, median, DESeq2 scaling factors, or RPKM/FPKM/TPM which normalize by gene length and reads mapped.
ML / AI: MetaVelvet-DL (Liang and Sakakibara 2021)
- Extension of the MetaVelvet assembler
- Uses deep learning to more accurately identify and partition de Bruijn graphs
- Improves genome assembly and resolution of individual species within complex microbial communities
Exploratory data analysis
- Methods like Principal Component Analysis (PCA), t-SNE, and UMAP are employed for dimension reduction and visualization
- Visualize global gene expression patterns
- Cluster samples with similar profiles
Summary
Metagenomics (16S or shotgun): characterization of entire microbial communities living in specific environments
Example
- Environment: Human skin
- Sample: Skin swabs are taken from the faces of many individuals
- 16S approach: Characterization of microbial diversity on human faces
- Shotgun metagenomics: Characterization microbial diversity and functional / genetic diversity of microbes on human faces
Future Directions
- Whole-genome sequencing (WGS) data is increasingly used for classification and identification
- Single-cell methods are being developed for metagenomics
- Ongoing improvement of sequencing platforms (like Nanopore) and computational tools (like deep learning assemblers) continues to drive the field forward