Creation of these notes included brainstorming and proof reading with Gemini.
Introduction to Bioinformatics
Bioinformatics is essentially big data analysis for biological data sets. It requires computational and statistical analyses in order to extract meaning from biological data. Since this is the case, bioinformatics can also be used to refer to the development of the software and methods which are used to understand biological data. Bioinformatics is an interdisciplinary field where biology, computer science, and statistics meet. (Genomics 2021)
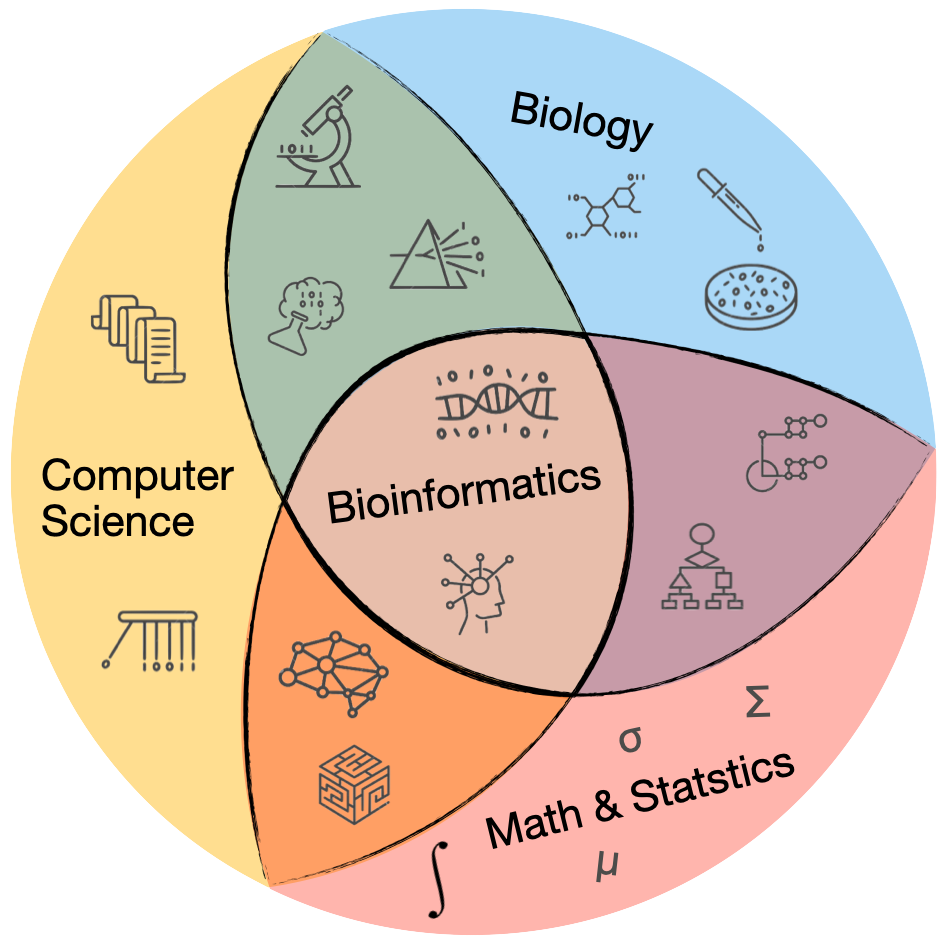
Bioinformatics is an interdisciplinary field
Bioinformatics sits at the intersection of biology, computer science, mathematics and statistics.
Bioinformatics is in a constant state of growth
The field has experienced rapid development due to the exponential increase in both sequence information accumulated in databases and available computer processing power.
Why is Bioinformatics Important?
Some answers could include:
It supports the transition of biology into a data-driven science. This is especially important with advancements in technologies like next-generation sequencing and mass spectrometry.
Bioinformatics is essential for processing, analyzing, and interpreting the massive amounts of biological data generated, including genetic information, protein amino acid sequences, and protein structures.
It helps organize, retrieve, store, and integrate diverse protein data from various sources, making it accessible for comprehensive analysis.
Bioinformatics facilitates a deeper understanding of cellular mechanisms, disease progression, and the intricate relationship between genotype and phenotype.
The Central Dogma of Molecular Biology
Original concept: The central dogma of molecular biology is a theory stating that genetic information flows only in one direction (Crick 1958):
DNA transcription to RNA
RNA translation to protein
This has become a little less strict as we’ve come to recognize some exceptions to this rule. For example, some viruses contain only RNA. Also, prions are infectious proteins that have the ability to replicate without DNA or RNA. Some examples include:
During transcription, RNA polymerase attaches to the DNA strand at a promoter. RNA polymerase is often assisted by transcription factors, that help recruit RNA polymerase to specific promoter regions.
Translation
During translation, transcribed mRNA enters the cytoplasm and is translated into protein, one codon at a time, inside of a ribosomal complex until a stop codon is encountered on the mRNA strand.
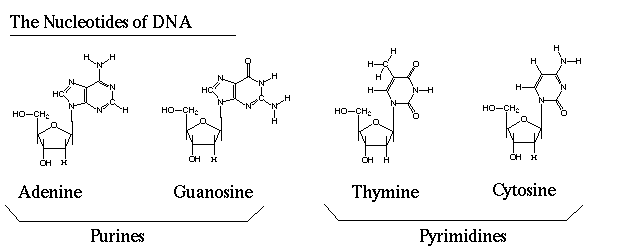
Codons
As seen above, each triplet of nucleotides (a codon) specifies a particular amino acid, which are the fundamental building blocks of proteins. Side note: thymine is transcribed to uracil (U) when synthesizing mRNA.
The prediction of protein sequences from DNA sequences in bioinformatics is based on this dogma
Information flows from DNA to protein, not the other way around.
While precise functional prediction is possible for only about one-third of coding genes from bacterial genomes (Price et al. 2018), bioinformatics tools can provide strong hypotheses for further experimental validation.
These amino acid sequences twist and fold into proteins that perform a multitude of biological functions. Here is a protein structure of human CCR5:
https://www.rcsb.org/structure/4MBS
CCR5 is the primary entry point of the HIV virus when infecting T cells.
Genome annotation
One major task in bioinformatics is genome annotation:
Identifying and labeling relevant features
Predicting coding genes
Predicting structural genes like ribosomal RNA (rRNA).
This process often begins with identifying open reading frames (ORFs), which are stretches of DNA capable of being translated into amino acids without encountering stop codons.
Functional annotation
Functional annotation further connects these predicted genes to their biological processes by assigning Gene Ontology (GO) terms across three functional categories:
Molecular function
Biological processes
Cellular components
Biological Databases
Due to the size and complexity of biological data, specialized databases are necessary for organizing and interacting with data:
Storage
Management
Searching & retrieval
FAIR data
For reliability, databases must be continuously updated and their documentation easily accessible. They are expected to adhere to the FAIR principles, making data:
Findable
Accessible
Interoperable
Reusable
Data sources
There are hundreds of biological databases
Primary Databases: contain raw experimental data
Secondary Databases: are derived from primary data
Added interpretation
Quality control
Reduced redundancy
Primary database examples
GenBank: The most widely used and well-known primary database for DNA sequences. It is managed by the National Center for Biotechnology Information (NCBI). GenBank is an open-access, annotated collection of publicly available nucleotide sequences.
European Nucleotide Archive (ENA) / EMBL: primary database for DNA sequences in Europe, it is integrated into ENA and hosted by the European Bioinformatics Institute (EBI-EMBL) in the UK.
Sequence Read Archive (SRA): A primary database from NCBI for storing raw sequence reads generated by high-throughput experimental techniques.
Secondary databases
UniProt (Universal Protein Resource): A major secondary protein database that includes highly curated entries (known as “Swiss-Prot”) and computationally derived ones (TrEMBL). It offers comprehensive protein sequences, functional annotations, and structural/domain information.
Protein Data Bank (PDB): A repository for experimentally determined 3D structures of proteins and nucleic acids, typically obtained through methods like X-ray crystallography or NMR spectroscopy.
InterPro: An integrated resource that combines data from various sources (e.g., Pfam, PROSITE, PRINTS, SMART) to predict protein domains, families, and functional regions.
KEGG (Kyoto Encyclopedia of Genes and Genomes): A comprehensive database that integrates genetic, chemical, and functional information, providing pathway maps and functional annotations for genes and proteins.
Navigating and retrieving data
Accessibility: Most major bioinformatics databases like NCBI, UniProt, and PDB are freely accessible via web interfaces.
Search Mechanisms:
Users can search these databases using keywords or specific accession numbers, which are unique identifiers for sequences.
Many (most?) databases also have APIs that can be accessed using R and python.
FASTA format
A very common and simple text-based format for representing nucleotide or amino acid sequences.
First line of a sequence starts with >, followed by a sequence identifier/name
Subsequent lines contain the actual sequence
For example, here is an amino acid sequence for the human CCR5 gene.
The Basic Local Alignment Search Tool is the most frequently used bioinformatics program for comparing a query sequence (DNA or protein) against all sequences in a chosen database.
BLAST is a fast and reliable heuristic algorithm for finding regions of sequence similarity (homologs).
Its output includes a statistical value called the E-value, which is a measure of the probability that an alignment occurred purely by random chance.
Applications of bioinformatics in molecular biology
More than just data management - powerful computational methods to investigate
protein structures
protein-protein interactions
complex relationships between structure and function
Key Application Areas
Protein ID: processing mass spectrometry data to identify and quantify peptides and proteins.
Functional annotation and relationships: identify conserved domains, functional sites, and motifs based on sequence and structural analysis.
Gene Ontology (GO)
Drug discovery:
Virtual screening
Molecular dynamics simulations
Ligand docking
Prediction of mutation impact on therapeutic effectiveness
Disease diagnostics: analysis of diverse data types (e.g., clinical symptoms, laboratory test results, medical images, genomic sequences) to make accurate disease predictions.
Gene expression analysis: analysis of differentially expressed genes using RNA-Seq and single-cell RNA-Seq data analysis pipelines, which involve:
Quality control
Read mapping
Expression quantification
Differential analysis
Clustering / cell type inference
Reconstructing biological networks: reconstruction of intricate protein interaction and signaling networks.
Cytoscape is commonly used for visualizing and analyzing these complex networks
Future Directions
AI/ML integration
Multi-omics data integration
Challenges and opportunities
Interpretability of AI models
Data-hungry AI models
Amount of data
Quality of data
Expanding Scope
Collaboration
References
Crick, F. H. 1958. “On Protein Synthesis.”Symposia of the Society for Experimental Biology 12: 138–63.
Eng, Jimmy K., Ashley L. McCormack, and John R. Yates. 1994. “An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database.”Journal of the American Society for Mass Spectrometry 5 (11): 976–89. https://doi.org/10.1016/1044-0305(94)80016-2.
Price, Morgan N., Kelly M. Wetmore, R. Jordan Waters, Mark Callaghan, Jayashree Ray, Hualan Liu, Jennifer V. Kuehl, et al. 2018. “Mutant Phenotypes for Thousands of Bacterial Genes of Unknown Function.”Nature 557 (7706): 503–9. https://doi.org/10.1038/s41586-018-0124-0.